Amyotrophic Lateral Sclerosis (ALS)
https://www.istockphoto.com/photos/amyotrophic-lateral-sclerosis
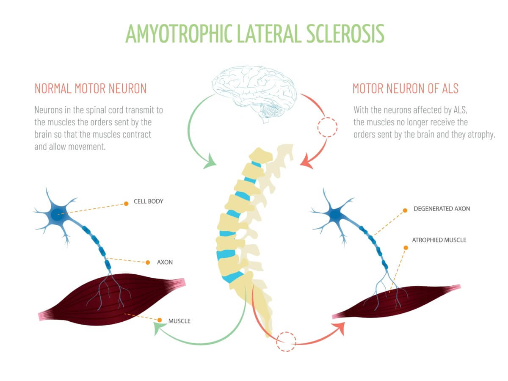
Neurodegenerative diseases are very difficult to treat. While treatments have been developed in an attempt to slow the progression of various neurodegenerative diseases, very few have been successful (1). One neurodegenerative disorder in particular that has been very difficult to treat is Amyotrophic Lateral Sclerosis also known as ALS, or Lou Gehrig’s disease. ALS is a disorder that causes the degeneration of motor neurons (cells that send signals from the brain to the muscles) within the motor cortex of the brain (2). The motor cortex is the area of the brain located at the front of the brain that helps you plan and carry out voluntary movements such as walking, writing, and speech (3). The motor cortex is made up of two main areas: the primary and nonprimary motor cortex. The primary cortex is responsible for initiating movements, while the nonprimary motor cortex plans the movements that are being initiated (3). The motor neurons located in the motor cortex communicate with other motor neurons within the spinal cord that allows muscle movements/contraction. The degeneration of the motor neurons within the motor cortex leads to the death of the neurons which leads to a loss of control of muscle movements, and eventually paralysis (3). While it is important to gain a scientific understanding of ALS through who is diagnosed, how symptoms appear, and how long it takes to receive treatment, this knowledge can’t capture the day-to-day reality of living with ALS. The physical changes, the emotional impact, and the challenges of getting a diagnosis all unfold in ways that data alone cannot fully explain which is why it is important to turn to the story of one patient whose journey with ALS reflects the complexities of diagnosis and progression.
Mrs. Tina Henson was a family friend who was diagnosed with ALS in 2025. I met her in 2019 through her son, Dylan Henson who was one of my childhood friends. Through my friendship with Dylan, I grew to know Mrs. Tina as a lively and heavily involved parent. We grew closer through random dinner dates at her home and family karaoke nights. During the summer of 2023 while on a Target run, I ran into her and noticed she was in a motorized cart. When I asked what was wrong, she mentioned that she was recently diagnosed with drop foot but that she was overall doing okay. From 2023 to 2025, her symptoms gradually worsened leading up to her passing on March 17th, 2026 at the age of 50. During her journey with ALS, she gave personal accounts of her experiences and intimate insights into what it was really like to live with the disease day-to-day.
Mrs. Tina Henson first started experiencing what was originally diagnosed as drop foot in her right foot in Summer 2023. She began going to physical therapy and continued from 2023 to 2024. While doing physical therapy she also started stem-cell infusions until she noticed muscle weakness in her left foot as well. After discontinuing the stem cell infusions, her muscle weakness progressed leading to her becoming fully paralyzed from the waist down in November 2024. She continued physical therapy following the partial paralysis until her physical therapist started to notice her condition worsen.
When patients start to experience symptoms, there are two main onset sites where symptoms may occur (4). When ALS first occurs in the muscles affecting speech and ability to chew and swallow, it is referred to as bulbar-onset ALS. When ALS occurs in the muscles primarily affecting the muscles in the arms and legs it is known as limb-onset ALS as seen in Mrs. Tina’s case (4). Limb-onset ALS has been found to be more common with bulbar-onset ALS only accounting for roughly 25% of patient cases (5). Median survival following the onset of symptoms is approximately 3 to 5 years with 30% of patients surviving 5 years. Studies have also shown that factors such as mild obesity at the time of diagnosis and having limb-onset ALS have improved survival rates (5). But, the first challenge is receiving a diagnosis.
With mixed emotions Mrs. Tina explained her experience with receiving a diagnosis: “I went to the doctor on Friday…I remember the physical therapist said…you might still want to go a neurologist because I don’t think this is muscular… just to be safe. I went out to the desk after my appointment to ask when I could see a neurologist as soon as possible. It was like next year… before I could see one, until the young doctor. I leave the doctor’s office, it’s a Friday… I went to another appointment on Monday to see the young neurologist. [He said] ‘before we name all of the 25 other things that it could be’, he asked if I had ever heard of motor neuron disease, and I was like ‘no’. He said ‘PLS and ALS’ and I was like what? This is when I was still walking… I was sitting in the doctor’s office and I was thinking… all this time the other doctors were saying it was my back. I remember crying and getting in the car and I didn’t even know where to drive. I drove to the lake and I cried and cried. So I looked it up and it said motor neuron disease also known as ALS… there are a thousand different reasons for foot drop. I hadn’t had one MRI, one CAT scan for him [the doctor] to say that.”
Following her diagnosis and paralysis from the waist down, her symptoms began to worsen. First, she started noticing differences in her breathing and low oxygen levels during doctors’ visits. In June of 2025, she received a breathing machine to assist with the weakness in her diaphragm. By August of 2025, she started to experience weakness and tremors in her arms, eventually spreading to her fingers by the end of November. By December of 2025, Mrs. Tina was paralyzed from the neck down.
“This disease is like a monster in my body… this a pain that I wouldn’t want anybody to feel. This disease took my legs, my arms, my fingers, my toes, my body. All I got is my voice and my voice is going. I can’t move my tongue in the morning… I’m coughing all night. I’m choking, I’m waking up… I feel like I’ve fought for three years.”- Tina Henson
To gain a more nuanced understanding of these symptoms and how ALS is diagnosed, it is important to look at the proposed underlying causes of how ALS affects the brain, and as a result, the body.
Proposed Causes of ALS
Unfortunately, the degeneration of motor neurons caused by ALS is not caused by one specific mechanism making it even more difficult to effectively treat ALS. There have been many hypotheses surrounding the mechanism of how motor neuron degeneration occurs including neuron overstimulation and genetic mutations (6). When neurons need to communicate to each other, they release chemicals called neurotransmitters (7). One of these neurotransmitters, glutamate, acts like a signal that tells the receiving neuron to “turn on”. When too much glutamate is present, it overstimulates the receiving neuron, like pressing a button too many times, which can damage or kill the neuron (7;6). This hypothesis suggests that ALS causes an increase in glutamate release, ultimately resulting in cell damage or death through the cellular mechanisms previously discussed. One study that sought to confirm this hypothesis tested patients with ALS for glutamate concentration using their cerebral spinal fluid (CSF), a clear liquid that surrounds and protects the brain and spinal cord, which found that patients with higher rates of muscle deterioration and more impaired limb function had higher concentrations of glutamate (8). However, 40.8% of the ALS patients in the study had normal glutamate concentrations, which suggests that further research is necessary to continue investigating the potential cellular mechanisms that underly ALS (8).
Another hypothesis for how ALS leads to motor neuron degeneration is through the mutation of specific genes leading to proteins clumping together instead of functioning properly (protein aggregation or accumulation). Now for moment, I want you to imagine you are baking a loaf of bread. It is very important to follow the recipe to get the desired result of a delicious, airy, well-grown loaf of bread. If you substitute eggs for yeast, you might still get something that resembles bread, it will be a very different result. Yeast is very important for the structure and the binding of the bread, and though eggs can create leavening, they will not produce a light airy texture. Similar to the yeast in the analogy, within the nucleus of neurons, there is a protein that is crucial for neuronal function (9). In patients with ALS, a gene (small sections of DNA strands) provides instructions to create the protein responsible for neuronal function (10; 9). There is a method that allows researchers to observe a mutation on the gene that provides the “incorrect” instructions that result in the accumulation of the protein leading to neuron degeneration (10).
One way to test for genetic mutations is through fluorescent DNA sequencing which allows researchers to read DNA by tagging it with glowing markers, which helps them spot small changes, or mutations in genes. DNA is made of building blocks that pair together in specific ways. If one of these blocks is changed, it can alter the resulting protein (7). The fluorescent markers will identify abnormalities within the DNA sequencing and potential nucleotide substitutions that lead to the production of different proteins that potentially damage the neuron (11). While the understanding of these genetic mutations is promising, they are not present in all patients with ALS, showing the need for further research into the causes of ALS.
Demographic Differences within ALS
In addition to the complex causes of ALS, there have also been differences in how different demographics of patients experience ALS and their time of diagnosis. An analysis of data from 4,242 patients with ALS collected from surveys from the National ALS Registry (12) observed differences in symptom onset, the specific types of symptoms experienced, BMI (Body Mass Index) prior to diagnosis, and more. Results showed that a significantly higher percentage of Black patients experienced limb-onset specifically in their arms/hands (12). White patients received an ALS diagnosis about 16 months after experiencing muscle weakness, while Black patients did not receive a diagnosis until approximately 24 months after their symptom onset (12). While the reasons behind these differences are not known, the results give insight into how ALS can affect different demographics in different ways. Not knowing how ALS affects different demographics can lead to misdiagnoses and delayed treatment. The timing of treatment in proximity to diagnosis was also analyzed between demographics (12). Patients that were not identified as Black or White used breathing support devices 11 months prior to their diagnosis, while White patients used the equipment 1 month prior (12). Black patients, however, reported not using the breathing devices until 6 months after their diagnosis, which could have potentially led to a shorter survival duration (12) While understanding the demographic differences in symptoms and treatment and potential causes of ALS can lead to further development of treatments for ALS, remembering the people who are affected by the disease is necessary for wholistic care of patients with ALS.
Despite the internal battle that Mrs. Tina fought for three years, she still made continuous efforts to make those around her laugh and keep them uplifted. After all she had gone through, she still found a way to provide guidance and reflect on her own experience with ALS.
“That would be my message that I would want to tell someone, live your life, even if you don’t go to get tested [for ALS], go on a trip, walk with a cane, walk with a walker, walk with a limp…”- Tina Henson
The self-reports from the ALS registry and Mrs. Tina’s account show the importance and necessity of being knowledgeable of the various ways ALS can show up in different demographics of patients. Having this knowledge and listening to the experiences of patients can also help neurologists provide better treatment for patients and catch symptoms early. As mentioned earlier, having limb-onset can potentially impact survival rates for patients which is why it’s important for neurologists to be able to accurately and efficiently identify when a patient could potentially be suffering from motor neuron degeneration. Stories like Mrs. Tina’s should be heard to not only help provide better treatment for patients just like her, but also to potentially slow the progression of ALS allowing patients more time.
References
1. Gadhave, D. G., Sugandhi, V. V., Saurav Kumar Jha, Nangare, S. N., Gupta, G., Sachin Kumar Singh, Dua, K., Cho, H., Hansbro, P. M., & Keshav Raj Paudel. (2024). Neurodegenerative Disorders: Mechanisms of Degeneration and Therapeutic Approaches with Their Clinical Relevance. Ageing Research Reviews, 99, 102357–102357. https://doi.org/10.1016/j.arr.2024.102357
2. ALS Association. (2024). Disease Mechanisms. The ALS Association. https://www.als.org/research/als-research-topics/disease-mechanisms
3. Guy-Evans, O. (2021, February 15). Neuron Function, Parts, Structure, and Types. SimplyPsychology.org. https://www.simplypsychology.org/motor-cortex.html
4. Ravits, J., Appel, S., Baloh, R. H., Barohn, R., Brooks, B. R., Elman, L., Floeter, M. K., Henderson, C., Lomen-Hoerth, C., Macklis, J. D., McCluskey, L., Mitsumoto, H., Przedborski, S., Rothstein, J., Trojanowski, J. Q., van den Berg, L. H., & Ringel, S. (2013). Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 14 Suppl 1, 5–18. https://doi.org/10.3109/21678421.2013.778548
5. Brotman, R. G., Moreno-Escobar, M. C., Joseph, J., Sunil Munakomi, & Pawar, G. (2024, February 12). Amyotrophic Lateral Sclerosis. Nih.gov; StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK556151/?report=classic
6. Tolochko, C., Shiryaeva, O., Alekseeva, T., & Dyachuk, V. (2025). Amyotrophic Lateral Sclerosis: Pathophysiological Mechanisms and Treatment Strategies (Part 2). International Journal of Molecular Sciences, 26(11), 5240. https://doi.org/10.3390/ijms26115240
7. Hedges, V. (2022). Introduction to Neuroscience. In openbooks.lib.msu.edu. Michigan State University Libraries. https://openbooks.lib.msu.edu/introneuroscience1/
8. Spreux-Varoquaux, O., Bensimon, G., Lacomblez, L., Salachas, F., Pradat, P. F., Le Forestier, N., Marouan, A., Dib, M., & Meininger, V. (2002). Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. Journal of the Neurological Sciences, 193(2), 73–78. https://doi.org/10.1016/s0022-510x(01)00661-x
9. Xu, Z., & Yang, C. (2014). TDP-43—The key to understanding amyotrophic lateral sclerosis. Rare Diseases, 2(1), e944443. https://doi.org/10.4161/21675511.2014.944443
10. Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., Clay, D., Wood, E. M., Chen-Plotkin, A. S., Martinez-Lage, M., Steinbart, E., McCluskey, L., Grossman, M., Neumann, M., Wu, I-Lin., Yang, W.-S., Kalb, R., Galasko, D. R., Montine, T. J., & Trojanowski, J. Q. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. The Lancet Neurology, 7(5), 409–416. https://doi.org/10.1016/s1474-4422(08)70071-1
11. SNP Genotyping Analysis Using TaqMan Assays | Thermo Fisher Scientific – UK. (2019). Thermofisher.com. https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-assays/snp-genotyping-taqman-assays.html
12. Raymond, J., Nair, T., Kelly Graham Gwathmey, Larson, T., D. Kevin Horton, & Mehta, P. (2024). Racial Disparities in the Diagnosis and Prognosis of ALS Patients in the United States. Journal of Racial and Ethnic Health Disparities. https://doi.org/10.1007/s40615-024-02099-6
Excellent article!